Liverpool Superregional Craniofacial Team
Download the leaflet
Genetics of Scaphocephaly (Sagittal Synostosis) (411kB pdf)
Sagittal Synostosis
Your child has been referred to the Liverpool Supraregional Craniofacial Service; one of 4 highly specialised craniofacial services in England who will see and assess your child. They will discuss the options available for your child – these will depend on your child’s age, severity of the sagittal synostosis and parental and child choice. This service will also follow up your child into adolescence.
Introduction to Craniosynostosis
The developing skull is made from different bones which are separated by growth lines called sutures (Fig 1). These sutures allow the bones of the skull vault to overlap slightly so that the baby’s head can pass through the birth canal during delivery. The sutures also allow for the brain to grow rapidly during the first two years of life. If one or more of the sutures fuse too early this is called ‘craniosynostosis’. Depending on which suture is affected, there will be a characteristic change in head shape, and it may also influence how the face grows and develops.
Craniosynostosis is usually described in one of the following 2 ways:
1. ‘Non-syndromic’ where only one suture is involved and there are no other features. This is the most common type accounting for approximately 75% of all craniosynostosis. Sagittal synostosis most commonly is ‘non-syndromic’.
2. ‘Syndromic’ where more than one suture is fused and there are other physical features or difficulties that follow a recognisable pattern. Syndromic causes account for approximately 25% of all patients seen with craniosynostosis. There are a small number of syndromes that can have sagittal suture synostosis as a feature.
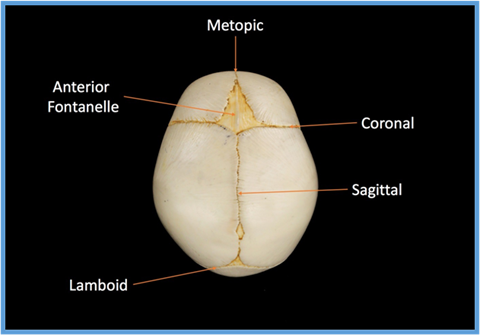

Sagittal synostosis occurs when the sagittal suture – the growth line that runs from front to back along the top of the skull (Fig 1) fuses too early. It is the most common craniosynostosis condition affecting a single suture. It happens approximately 1 in every 3000 births and accounts for 40-55% of all non-syndromic craniosynostosis seen. It affects males approximately four times more frequently than females for reasons that are not understood.
Scaphocephaly is the name given to the resulting head shape. The normal head shape is wide at the back, narrower at the front and slopes down towards the front. Individuals with scaphocephaly have a head shape that is wider at the front (this can make the forehead look prominent), narrower at the back and slopes in the opposite direction. It is sometimes described as looking like an upturned keel of a boat and this is the meaning of the word ‘Scaphocephaly’. There are variations in the head shape seen if the suture is partially fused or fuses in an atypical way.

In most individuals with sagittal synostosis this is the only finding and it is said to be ‘non-syndromic’. It is only part of a wider syndrome in a small proportion of patients. If there is no one else in the family with an unusual head shape, and if your child has no other medical problems or unusual features, then we would say this is likely to be non-syndromic sagittal synostosis.
Individuals with sagittal synostosis may have or develop:
1. An unusual head shape: Characteristically this is longer and thinner than average with a prominent forehead, bullet-shaped back of head and reduced height to the back of the skull. Variation includes having a saddle deformity with a lower area across the centre of the skull. There is often ridging over the fused suture. Some children will be more severely affected than others. The abnormal head shape is normally present at birth and can worsen for the first few months of life. At some point the head shape will stabilize but it is very unlikely to improve without treatment.
2. Raised intracranial pressure (ICP): This is thought to be due to a mismatch between the growth of the developing brain and the skull it is contained in causing increased pressure inside the skull. Other factors may contribute to this, however if raised pressure occurs it can damage vision and cause developmental delay. In sagittal synostosis it is unlikely to occur in children in
their first few years of life or in children over 10. The chance of developing raised pressure is likely to be around 20% over childhood but some scientific literature reports rates as high as 44%. Symptoms are headaches, especially in the morning and decline in rate of development. Intracranial pressure is monitored by a combination of ophthalmology checking the back of the eyes, scans, and clinical symptoms. In some patients the pressure is measured but this involves a small operation.
3. Speech and language delay – in the general population about 6-10% of children will have some impairment of their speech and language. Up to 37% of children who have sagittal suture synostosis experience delay in acquiring speech and language (Shipster et al. 2003). For this reason, the craniofacial teams have a specialist speech and language therapist who will monitor and assess your child and give advice to local speech and language therapists if necessary.
4. Psychological impact – sometimes families and young people have questions about their appearance and sometimes this can influence how they think, feel and behave. If these thoughts and feelings begin to impact on the family or child’s wellbeing, then support can be offered. Also, it is recognised that there is limited research on the learning abilities of children with a diagnosis of sagittal synostosis. Based on this, the craniofacial team will offer regular reviews/assessments of your child’s learning, as it is known that if it is monitored then support and advice can be given early to your child’s school and to you as a family. A clinical psychologist is also a member of the craniofacial team and will offer support and assessment.

At present we do not fully understand what causes the sagittal suture to fuse early. Many factors and including genetic contributions are likely to play a role. Recently a change in a gene called SMAD6 has been shown to predispose to sagittal synostosis approximately 1% of all patients with sagittal synostosis. It is now possible to offer testing of this gene and the craniofacial team will discuss this option with you. There is active research to understand these factors better and your craniofacial team will talk to you about research studies you could take part in.

The risk of craniosynostosis to any future children you may have is low as the majority are a ’one-off’ in a family. The risk is estimated to be around 14% and recurrence is most likely to involve the
sagittal suture but can involve one of the other sutures. The risk is highest for brothers of an affected individual.
In some rare families there is more than one affected family member and in this situation the recurrence risk may be higher. A genetics doctor working in the craniofacial team will be able to assess your risk more accurately in this situation and discuss genetic testing available.

References
Further reading
Shipster C, Hearst D, Sommerville A, Stackhouse J, Hayward R, Wade A. Speech, language, and cognitive development in children with isolated sagittal synostosis Developmental Medicine & Child Neurology 2003, 45: 34–43
Timberlake AT, Choi J, Zaidi S, et al. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Ginsburg D, ed. eLife. 2016;5:e20125. doi:10.7554/eLife.20125.
Garza RM, Khosla RK. Nonsyndromic Craniosynostosis. Seminars in Plastic Surgery. 2012;26(2):53-63. doi:10.1055/s-0032-1320063.
Greenwood J, Flodman P, Osann K, Boyadjiev SA, Kimonis V. Familial incidence and associated symptoms in a population of individuals with nonsyndromic craniosynostosis. Genetics in medicine : official journal of the American College of Medical Genetics. 2014;16(4):302-310. doi:10.1038/gim.2013.134.
This leaflet only gives general information. You must always discuss the individual treatment of your child with the appropriate member of staff. Do not rely on this leaflet alone for information about your child’s treatment. This information can be made available in other languages and formats if requested.
Alder Hey Children’s NHS Foundation Trust
Alder Hey
Eaton Road
Liverpool
L12 2AP
PIAG 052